Cancer May Be Driven by DNA Outside of Chromosomes
In the last decade, researchers have come to realize that tumors harbor bits of extrachromosomal DNA that can drive malignancy.
Paul Mischel
Apr 1, 2021
338
In the spring of 2012, my colleagues and I began to notice something strange in tumor cells from patients with glioblastoma, a highly aggressive form of brain cancer, who were coming into our clinic at the University of California, Los Angeles. From genomic sequencing of their tumors, we knew they displayed amplification of a specific growth-promoting oncogene. Despite being treated with drugs designed to target this gene, the patients were not getting better, and when we interrogated the genomes of their cancers after the tumors were surgically removed following treatment, we saw that they had changed. The tumors had dramatically reduced the number of copies of the targeted epidermal growth factor receptor (EGFR) gene, presumably giving them an advantage to escape the drugs, and they had evolved these genetic differences at a rate that seemed to make no sense—within just one to two weeks.
Normally, we think of cancers evolving over many cell divisions, as the cells carrying genetic changes that provide a fitness advantage—such as an ability to resist a particular treatment—will be more likely to survive and divide. Here, we were noticing a change in the copy number of the gene within just a few generations. There was no way that we could explain how the tumors were altering their DNA so quickly.
Even stranger, we could take any cell from the tumor, and whether it had high or undetectable protein levels of EGFR, it would give rise to a new tumor when cultured in the lab or implanted into a mouse. Each of these new tumors would then display the full spectrum of cells found in the original tumor, varied in their EGFR copy number. This makes no sense according to what we know about classical genetics. We would have expected that tumors arising from a cell with low levels of EGFR would give rise to a tumor with low EGFR levels, whereas a tumor arising from a cell with high levels of EGFR would give rise to a tumor with high EGFR levels.
Extrachromosomal DNA was once thought to be a rare event in cancer, but our group showed that ecDNA is very common indeed.
In 2012, in a project spearheaded by a graduate student in my lab named David Nathanson, who is now an associate professor at the University of California, Los Angeles, we set out to understand why. We wanted to see where copies of EGFR were located. Typically when we sequence cancers, we grind up a tumor and “read” all of the genes present, looking for mutations and copy number variations, and then we assign the location of these genetic alterations to the chromosomes where those genes are in the human reference genome. But when we looked at a cell getting ready to divide—the only time when it’s possible to tell where a specific piece of DNA is located—we were surprised to find that EGFR was not where we’d predicted. In fact, it was not sitting on a chromosome at all. Rather, all of the amplified oncogenes were found on circles of extrachromosomal DNA (ecDNA). We could see these extra pieces of DNA near the chromosomes inside the nucleus of cancer cells.

Scanning electron microscopy images of lysed colon cancer cells reveal extrachromosomal DNA (small circles) in addition to chromosomes (larger, X-shaped structures). (Large spots are intact nuclei from other cells.)
KRISTEN TURNER, PRINCIPAL SCIENTIST AT BOUNDLESS BIO; PHOTOGRAPH TAKEN WHEN SHE WAS A MEMBER OF THE MISCHEL LABORATORY
When we removed the treatment with the EGFR inhibitor from cultured tumor cells, EGFR copy number quickly rebounded, but again, not on chromosomes. When we saw this, we realized that ecDNA might explain why some cancers can become resistant to treatment so quickly, allowing tumors to evolve at a rate that far exceeds anything that could be accounted for by classical genetics. We published our results in Sciencein 2014, but they were not immediately accepted by the community. Although we had only studied one tumor type, glioblastoma, we began to wonder whether this might be the tip of the iceberg.
Without realizing it, this study led us, and now others, to a series of discoveries that have changed the way that researchers view cancer in general, revealing frightening ways that tumors can evolve. We have learned that ecDNA is central to the behavior of some of the most aggressive forms of cancer, enabling remarkably elevated levels of oncogene transcription, creating new gene regulatory interactions, and providing a powerful mechanism for rapid change that can drive very high oncogene copy numbers or allow cancer cells to resist treatment.
Along the path of discovery, we found that we were not the first to have seen these extrachromosomal particles. But with new tools in hand, and new questions in mind, we saw them in a very different way. And when we took the time to peer deeply into the nucleus of cancer cells, we saw ecDNAs in abundance. We eagerly pursued studies to understand their importance in cancer progression and drug resistance, and even founded a biotech company, Boundless Bio, to identify and develop new ways to treat patients whose cancers are driven by ecDNA.
Misleading maps
Humans depend on maps to navigate complex landscapes and to understand the world around us. Without our maps, we’re lost. However, our maps also limit us. If something is missing from the map—or even worse, the map is wrong—we can be thrown off course. Consider Ptolemy’s and Copernicus’s maps of the solar system. Ptolemy made precise measurements of the planets moving across the heavens, but he got the orientation of his map wrong by placing Earth in the center. It took nearly 1,500 years for Copernicus to correct it.
Because ecDNAs are unevenly distributed between daughter cells, they drive variation among the cancerous cell population, fueling natural selection.
See full infographic: WEB | PDF
© EMMA CHENG
Researchers have been making maps of cancer for a long time. First we made organ-based maps, then cell type–based maps. These are still the dominant maps researchers and physicians use to navigate cancer, but the advent of powerful DNA sequencing technologies has ushered in a new era of cancer mapping at the level of the genome. We can take DNA from a patient’s tumor and find genetic mutations that are to blame. This has become the basis for cancer diagnosis and treatment. But, as we learned in our seminal study several years ago, something was missing from genome-based maps: ecDNA.
Since 1965, scientists have recognized that ecDNA elements exist in cells of certain cancers, in particular neuroblastoma. These elements were initially referred to as “minute chromatin bodies,” and then as “double minutes” because they were often paired. In the late 1970s and 1980s, a number of research teams showed that these double minutes could lead to gene amplification. In contrast with this earlier work, we found that ecDNAs are not always found in pairs. In fact, ecDNAs occur as paired bodies only 30 percent of the time. Therefore, for clarity, here we use the term “ecDNA” to describe both singlet ecDNA particles and double minute ecDNA.
Until very recently, despite efforts to decipher ecDNA structure, the importance of these ecDNA particles remained unclear, and as the field developed new techniques to generate genome information–rich maps of cancer, most researchers stopped thinking about ecDNA, which was thought to occur rarely. Indeed, according to the Mitelman database of chromosomal aberrations, ecDNA only occurred in about 1.4 percent of cancers. Thus, after we published our 2014 study showing that ecDNA is a very common event in cancer, there was what I would call a colossal scratching of heads. I would give talks about the work and people would say, for example, “That’s funny, the cancer-causing gene that I am studying is on chromosome 8.” I would then ask how they knew that, and they would present me with a copy of a cancer map based on the human reference genome. I realized that we might be looking at a “Ptolemy” map.
How ecDNAs Might Drive Cancer EvolutionIn contrast to chromosomal DNA, which is replicated and divvied up equally among daughter cells during cell division, the extrachromosomal DNA (ecDNA) found in some cancer cells is not always split evenly. Lacking centromeres, these circular bits of DNA are often unevenly parceled to daughter cells. Some daughter cells receive more ecDNA, which they then duplicate, so the copy number of oncogenes in those cells rises quickly. Moreover, because each cell division is essentially a “coin flip” with regard to ecDNA inheritance, variation among the cancerous cell population is preserved, providing an ample supply of the fuel needed for natural selection. These two features in combination could enable cancers containing ecDNA to evolve much more rapidly than cancers lacking ecDNA can. |
 © EMMA CHENG |
Appreciating extrachromosomal DNA
After our Science paper was published, I began working very closely with Vineet Bafna, a computer scientist at the University of California, San Diego, who had played a critical role in decoding the human genome. In a study spearheaded by a researcher from my lab named Kristen Turner (now at Boundless Bio), along with computer scientists Viraj Deshpande and Doruk Beyter from Bafna’s group, we integrated the old with the new—modern genomic tool kits, powerful next-generation sequencing, advanced computational methods, and visualization under the microscope of the locations of ecDNA. This work revealed the presence of ecDNA in almost every cancer type we looked at. We further found that ecDNA enabled cancers to change their genomes quickly, and even to move genes back onto chromosomes, but not in their original locations.
In early 2017, I gave a seminar on these findings at Stanford University, where I met Howard Chang, a physician-scientist there who had developed techniques for analyzing the chromatin architecture and epigenetic structure of cancer cells. Chang had recently discovered major changes in the structure of chromatin in cancer that allow transcriptional machinery greater access to the DNA to drive transcription. After my talk, we discussed how ecDNA might generate a far more open chromatin pattern, enabling enhanced transcription of oncogenes, in line with what Chang was finding. Working closely with Chang, as well as with Bafna and the University of California, San Diego’s Bing Ren, my postdoctoral fellows Turner, Sihan “Sean” Wu, and Nam Nguyen set out to better understand the structure of ecDNA and its transcriptional consequences.

Extrachromosomal DNA is spotted in this false-colored scanning electron microscopy image of lysed colon cancer cells. (Large spots are intact nuclei from other cells.)
KRISTEN TURNER, PRINCIPAL SCIENTIST AT BOUNDLESS BIO; PHOTOGRAPH TAKEN WHEN SHE WAS A MEMBER OF THE MISCHEL LABORATORY
We first demonstrated that ecDNA is circular. These ecDNAs differ from the more common small extrachromosomal circular DNA particles (eccDNAs) that are found in the genomes of eukaryotes from yeast to flies to humans. For one, eccDNAs average only about 100–1,000 base pairs in length and are thus relatively unlikely to contain intact genes or other functional elements, while ecDNAs are relatively large, averaging around 1.3 million base pairs. ecDNAs are chock-full of growth-promoting oncogenes, and they contain other genes and regulatory regions that may be involved in tumor formation and progression.
Moreover, unlike other types of small circular DNAs such as eccDNAs and ribosomal DNAs that can be found in normal cells at low levels, ecDNAs are unique to cancer, and they are highly amplified. And, because like all circular DNAs they lack the centromeres used to segregate homologous chromosomes, they are often not evenly divided between daughter cells upon cell division. This allows cancers with ecDNA to evolve rapidly, accumulating a great many copies of cancer-causing genes or reducing the number of drug-targeted oncogenes, while maintaining the cell-to-cell variability that drives accelerated tumor evolution and drug resistance. (See illustration above.)
The three-dimensional structure of ecDNA also holds clues to its role in some cancers. By combining ultrastructural microscopy and whole-genome sequencing with computational reconstruction and long-range DNA optical mapping in which very long fragments of DNA, sometimes more than 150,000 base pairs, can be resolved, my collaborators and I showed that ecDNAs are circular and that the DNA is wound around histone cores into nucleosomes like chromosomal DNA is, but in a highly abnormal fashion. ecDNA chromatin displays a significantly lower degree of compaction compared with the same DNA segments that reintegrate into chromosomes, suggesting that altered DNA shape may actively contribute to the transcription of oncogenes found on ecDNA. (See illustration below.) In fact, single-cell imaging further revealed that ecDNA is among the most transcriptionally accessible chromatin in the cancer genome in actively cycling tumor cells.
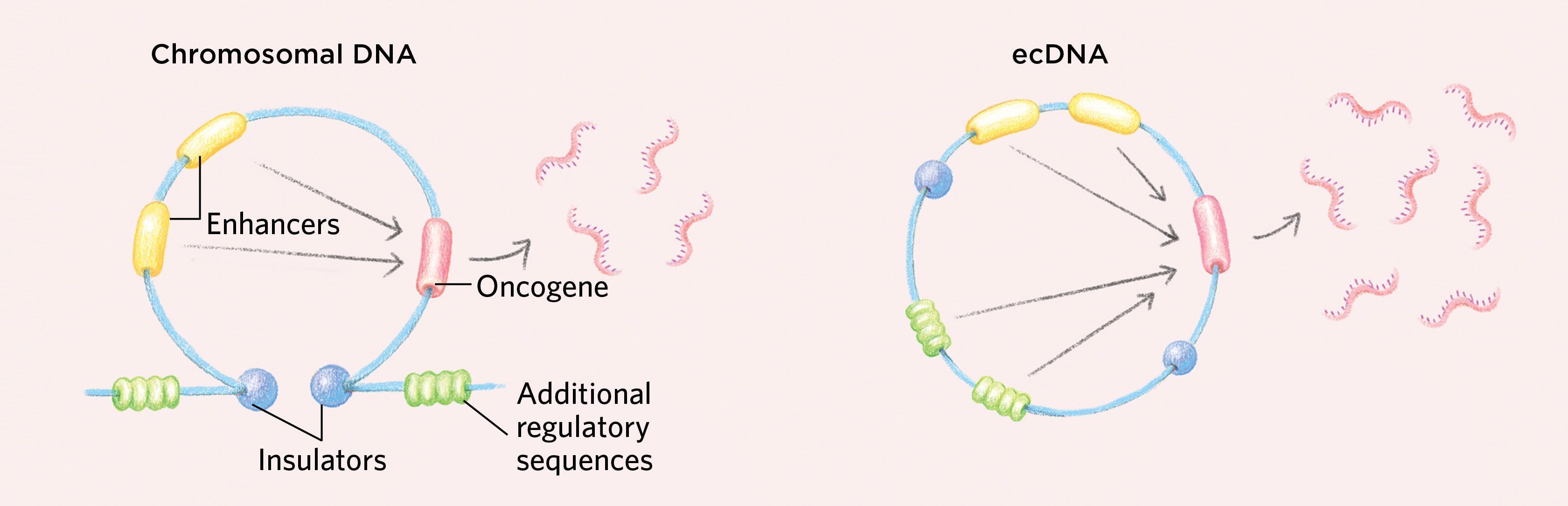
How ecDNAs Might Support Cancerous GrowthThe circular nature of ecDNAs can enable gene interactions that may support the increased transcription of oncogenes, as genetic elements normally found in distant parts of the genome may come together to interact. While insulators in the chromosomal DNA sit at the stem of a loop structure and ensure that regulatory sequences such as enhancers work only on their nearby target genes, the circular shape of ecDNA generates new interactions with additional regulatory sequences that would not normally occur on chromosomal DNA.
Additionally, ecDNAs tend to have a more-open chromatin structure than chromosomes that promotes increased gene expression. DNA is wound around histone cores into units of organization called nucleosomes. On chromosomes, some regions can become highly compacted, rendering the DNA inaccessible to the transcriptional machinery, but ecDNAs have an altered chromatin structure in which the nucleosomes do not compact, resulting in highly accessible DNA that is primed for transcription. Moreover, ecDNAs are loaded with active histone marks but have a paucity of repressive histone marks, promoting high levels of transcription.
|

We also found that ecDNA is loaded with chromatin modifications that promote transcription and has a paucity of repressive chromatin marks, suggesting that it is poised for high levels of gene expression. Further, we found that ecDNA chromatin is well organized into loops that are normally an important part of gene regulation, but with a three-dimensional topology that is distinct from that of chromosomal DNA. As the DNA segment becomes circular, in a process that is still incompletely understood, distal DNA elements are brought into proximity, enabling ultra-long-range chromatin interaction that cannot be achieved by chromosomal DNA. This could potentially form new gene regulatory circuits, including new active contacts that drive oncogenic transcription. Consistent with these findings, we found that oncogenes residing on ecDNA are in the top 1 percent of the most-transcribed genes in cancer cells that have them.
We published these results in Nature in November 2019, and the paper was highlighted in a story by Carl Zimmer in The New York Times. Very shortly thereafter, two other groups—one led by Peter Scacheri of the Cleveland Clinic and Case Western Reserve University and Jeremy Rich of the University of California, San Diego, and another by Anton Henssen of Charité Hospital in Berlin and Richard Koche of Memorial Sloan Kettering Cancer Center—added further evidence that ecDNAs may play a pivotal role in reorganizing the transcriptional control of cancer genomes by bringing regulatory elements encoded on ecDNA into contact with genes with which they would never interact in chromosomes.
Recent work from Chang, in collaboration with myself, Bafna, and Henssen, has begun to suggest a very exciting new way that these circular pieces of DNA, instead of acting alone, often organize themselves into nuclear bodies called ecDNA hubs. These hubs are tethered by proteins and appear to provide a platform for cooperative transcription, in which ecDNAs work together to drive the expression of cancer-promoting genes.
The question then became, how do we capitalize on this new understanding of ecDNA to improve patient outcomes? In 2018, Chang, Bafna, and I, with other scientists, cofounded Boundless Bio, where several staff scientists now seek the answer to that question.
Translating ecDNA to clinical application
Working closely with Chang, Bafna, and Roel Verhaak of the Jackson Laboratory (also a co-founder of Boundless Bio), we are trying to understand some of the clinical implications of ecDNA. Publicly available databases, including The Cancer Genome Atlas and the Pan-Cancer Analysis of Whole Genomes, contain a large number of whole-genome sequences of cancer samples, yielding a golden opportunity for discovery. We applied the AmpliconArchitect, a tool developed by Bafna that looks for the telltale signs of ecDNA in whole-genome sequencing data, including amplified regions that map to a circle, and then uses algorithms that deconvolute these circular structures. This enabled us to analyze the frequency and potential structural composition of ecDNA in more than 3,200 cancer samples of a wide range of histological types alongside matched whole blood and normal tissue. Our findings indicated that ecDNA is unique to cancer, and that at a minimum, 14 percent of human tumors, including some of the most malignant forms of cancer, harbor ecDNA.
Researchers have been making maps of cancer for a long time, but we now know that we’ve been missing something from our maps.
Further, we found that patients whose cancers have ecDNA have significantly shorter survival than cancer patients whose tumors are driven by lesions in chromosomal DNA. It remains to be seen how commonly ecDNAs play a role in the evolution of drug resistance, as we saw hints of in our initial study. Many other questions remain as well. Recent studies have shed light on how ecDNA may form, although we and others strongly suspect that there may be multiple routes to its development.
The problem of ecDNA in cancer, and the challenge that it represents, has become clear. The National Cancer Institute and Cancer Research UK recently designated ecDNA as one of the Cancer Grand Challenges that must be addressed. It is exciting to see mounting interest and an influx of talented investigators aiming to decipher the key aspects of ecDNA biology. We look forward to the development of new tools, new collaborations, and new treatments for patients.
Joshua Lederberg wrote in his landmark 1952 paper in Physiological Reviews: “I propose plasmid as a generic term for any extrachromosomal hereditary determinant.” In bacteria, circular plasmids are a powerful mechanism for gaining selective advantage because they enable rapid evolution, including drug resistance. Similarly, yeast, weeds, and even parasites can evade drugs and environmental toxins by encoding resistance genes on circular extrachromosomal DNA. ecDNAs may do the same for cancer, providing a potent vehicle for rapid tumor evolution that maximizes critical oncogenic gene variants—or reduces them to evolve drug resistance.
Just as explorers rely on maps of the Earth, and astronomers on maps of the galaxy, cancer biologists depend on maps to navigate the complexities of cancer. We now know that we’d long been missing a critical element. So here we are once again, as physiological cartographers, rolling up our sleeves and making new, topographically informed maps of cancer to help us navigate the multifarious disease and develop new and more effective treatments for patients.
Paul Mischel is a professor and Vice Chair for Research for the Department of Pathology at Stanford University School of Medicine and an Institute Scholar in ChEM-H at Stanford University.