Position Paper
Chondrosarcoma: Making the right diagnosis, grading and the proper classification
John M. Gross, MD
The Johns Hopkins Hospital
Baltimore, MD
Introduction
President’s remarks:Chondrosarcoma (CS) is a bone and soft tissue cancer that develops in cartilage cells particularly in the arm, shoulder, pelvis, knee, femur, spine, ribs and base of the skull. It is the second most prevalent primary malignancy of the bone. The prevalence of Chondrosarcoma is rare: estimates range from 1 to 5 in 1,000,000 people, depending on location. This wide range might be expected, based on the difficulties inherent in diagnosing CS, and the skills and equipment needed to do so. One would expect that the best staffed and equipped locations would have larger numbers than those who are not so fortunate, but this is not always the case.
Because Chondrosarcoma is rare, there is a lack of information. Primary care physicians and oncologists have little or no guidance on where to send patients with Chondrosarcoma. In the Summer, 2020 the Bone Cancer Research Trust (BCRT) conducted an international survey and reported that 65% of all patients with Chondrosarcoma are misdiagnosed. This results in significant delays in proper care and significantly impacts the outcome. This position paper contains diagnostic/pathological information geared to guide oncologists and other medical professionals to make appropriate referrals to pathologists that specialize in diagnosing rare bone and soft tissue sarcoma and to ensure the diagnosis is accurate the first time. The information in this position paper can also be very useful for CS patients to gain a greater understanding of their own diagnosis and can help them take charge of determining their specific course of treatment.
There are different types of Chondrosarcoma along with the differences in their grading (for example Conventional Chondrosarcoma ranges from Grade I/ good prognosis to Grade III/ poor prognosis). This position paper examines the challenges in diagnosing and classifying the different types and grades of Chondrosarcoma. Making the right diagnosis, classifying the right type and grade of Chondrosarcoma are critical to care and outcome.
For medical professionals this document is intended as an overview of the types and grades of Chondrosarcoma to familiarize you with this rare bone and soft tissue cancer. For patients and caretakers undergoing the diagnostic process I would make the following suggestions in your review:
1. Read the article carefully with particular interest to your diagnosis.
2. Use search engines to look up any medical terms that you do not understand.
3. Insist on getting your own pathology report and pictures of the slides from your biopsy.
4. Insist on having an opinion from an expert sarcoma pathologist who specializes in bone and soft tissue cancers about your diagnosis.
5. Find a Qualified Sarcoma Center with a multidisciplinary team to explore your treatment options.
6. Do not hesitate in seeking second opinions along the way, especially at crucial decision-making points (biopsy/pathology, treatment options, etc.)
7. Feel free to e-mail the Chondrosarcoma Foundation with any questions you have regarding the article and how it relates to your diagnosis. Our e-mail address is: info@csfshayna.org
Overview of Chondrosarcoma
Chondrosarcomas are malignant mesenchymal neoplasms (soft tissue tumors) of bone which produce cartilage matrix. Excluding myeloma, chondrosarcomas are the second most common primary malignant bone tumor after osteosarcoma, accounting for approximately 10% of all primary bone sarcomas. Chondrosarcomas are generally a disease of older individuals with a peak incidence in the 5th to 7th decade with a 2:1 male predominance. Pain is the most common clinical symptom and common sites for chondrosarcomas include the long bones, pelvic girdle, sternum/ribs and base of skull whereas chondrosarcomas are quite rare in the short tubular bones of the hands and feet.1,2
Chondrosarcomas may exhibit a wide range of behavior. Generally, chondrosarcomas are slow growing and relatively indolent but a subset show aggressive behavior and are associated with distant metastasis and poor outcomes. Therefore, the role of the pathologist is critical in properly grading and the classification of chondrosarcomas as histologic grade and subtype greatly dictates treatment and predicts behavior and prognosis. The purpose of this position paper is to review the challenges in the diagnosis of chondrosarcoma from the perspective of a pathologist with special attention on chondrosarcoma variants.
Conventional Chondrosarcoma
The vast majority of chondrosarcomas (roughly 80-90%) are known as conventional intramedullary (central) and occur as a primary neoplasm (tumor) in previously normal bone.1,2 Most conventional chondrosarcomas grow slowly and either do not metastasize or do so only late in their natural history. Conventional chondrosarcoma generally occurs in older patients within the center (medullary canal) of long bones of the extremities or within bones of the axial skeleton (pelvic bones, skull base, ribs, and sternum). Conventional chondrosarcoma is graded on a scale from I-III (low-grade: G1 to high-grade: G3). Grade I chondrosarcoma, synonymous with central atypical cartilaginous tumor when located in an extremity, is a hyaline cartilage neoplasm consisting of sheets of chondrocytes which show some features of aggressive behavior with the surrounding normal (host) bone either by histologic evaluation or by imaging.
However, from a nuclear cytologic standpoint, the distinction between an enchondroma (benign hyaline cartilage neoplasm that arises centrally within the medullary cavity of bone) and a low-grade (grade I) chondrosarcoma is generally thought to be one of the more difficult problems in bone tumor pathology. Figure 1 nicely shows a hyaline cartilage neoplasm completely surrounding (imprisoning) normal (host) bone suggesting aggressive behavior. A very common diagnostic scenario and challenge for a pathologist occurs with the receipt of numerous fragments of well-differentiated (low-grade) hyaline cartilage from a surgical curettage procedure in which the surrounding normal tissue is not present to assess for invasion (aggressive behavior). While radiologic correlation is always important in the diagnosis of bone tumors, in the setting of a low-grade hyaline cartilage neoplasm, radiology is essential. For example, figure 2 represents a CT scan of an intermediate-grade chondrosarcoma of the skull base presenting as an overtly aggressive mass. However, even with radiologic correlation, some cases of low-grade hyaline cartilage neoplasms can be extremely challenging (if not impossible) even amongst expert bone pathologists. Grade II conventional chondrosarcomas contain neoplastic hyaline cartilage that is obviously more cellular than normal cartilage and contains greater cellularity and nuclear atypia than what is acceptable for a grade I chondrosarcoma (Fig. 3). Grade II chondrosarcomas often show myxoid changes and have the ability to metastasize (Fig. 3). Greater than 90% of conventional chondrosarcomas are grade I (low-grade) or grade II (intermediate-grade) tumors; however, grade III chondrosarcomas are occasionally seen and show overt nuclear atypia, mitotic activity, spindled nuclei, tumor necrosis and have a high metastatic rate (Fig. 4). A large study by the Mayo Clinic revealed fewer than 5% of conventional chondrosarcomas are grade III.2
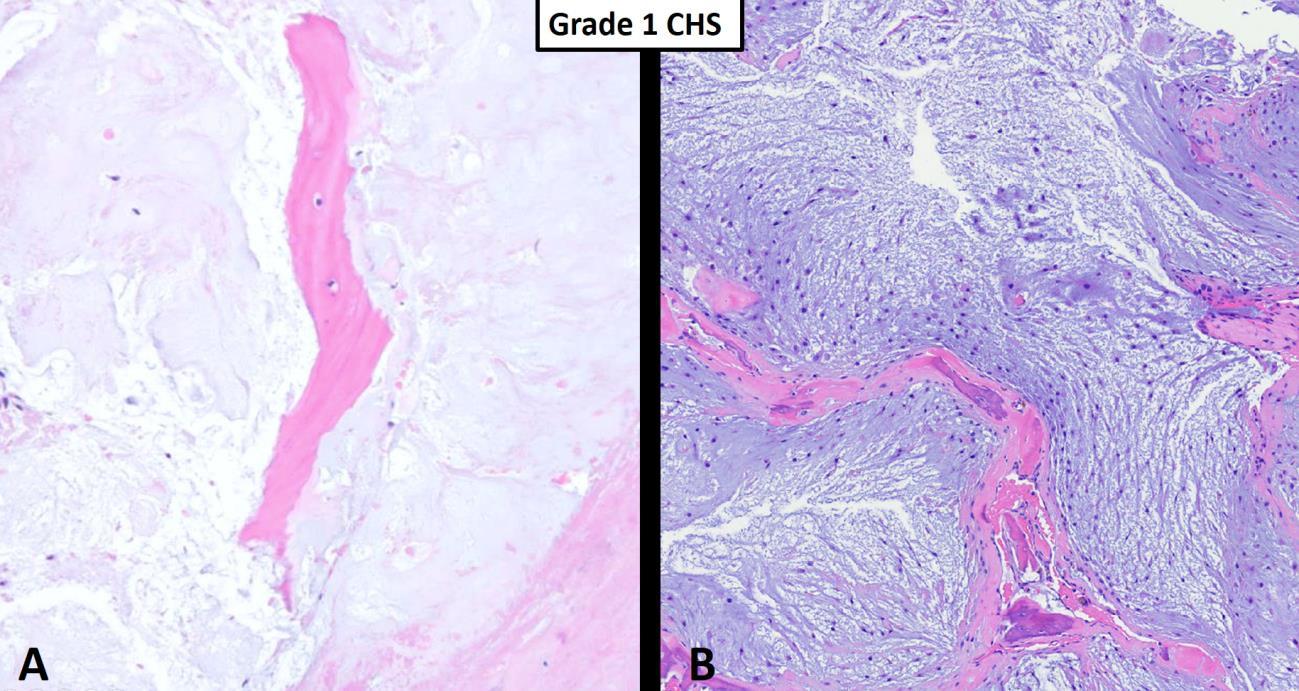
Fig. 1 Grade I chondrosarcoma. A Hypocellular hyaline cartilage neoplasm completely encasing pre-existing host bone (pink). B This intermediate power shows a grade I chondrosarcoma consisting of relatively hypocelluar hyaline cartilage neoplasm with myxoid changes permeating marrow cavity and entrapping host bone (pink). The host bone shows some remodeling changes suggesting a long standing, slow growing process consistent with a low-grade chondrosarcoma.
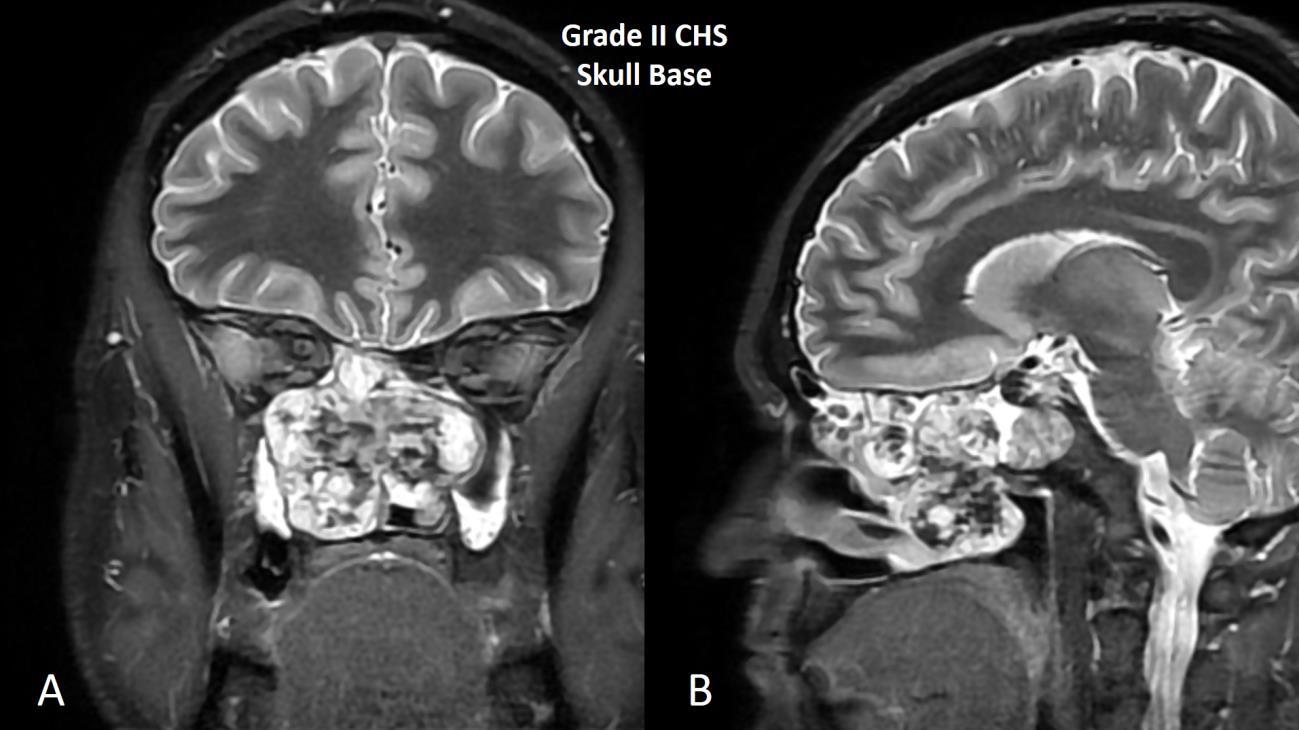
Fig. 2 MRI of grade II chondrosarcoma of skull base. A Coronal and B sagittal images shows a T2 bright destructive grade II chondrosarcoma of the skull base.
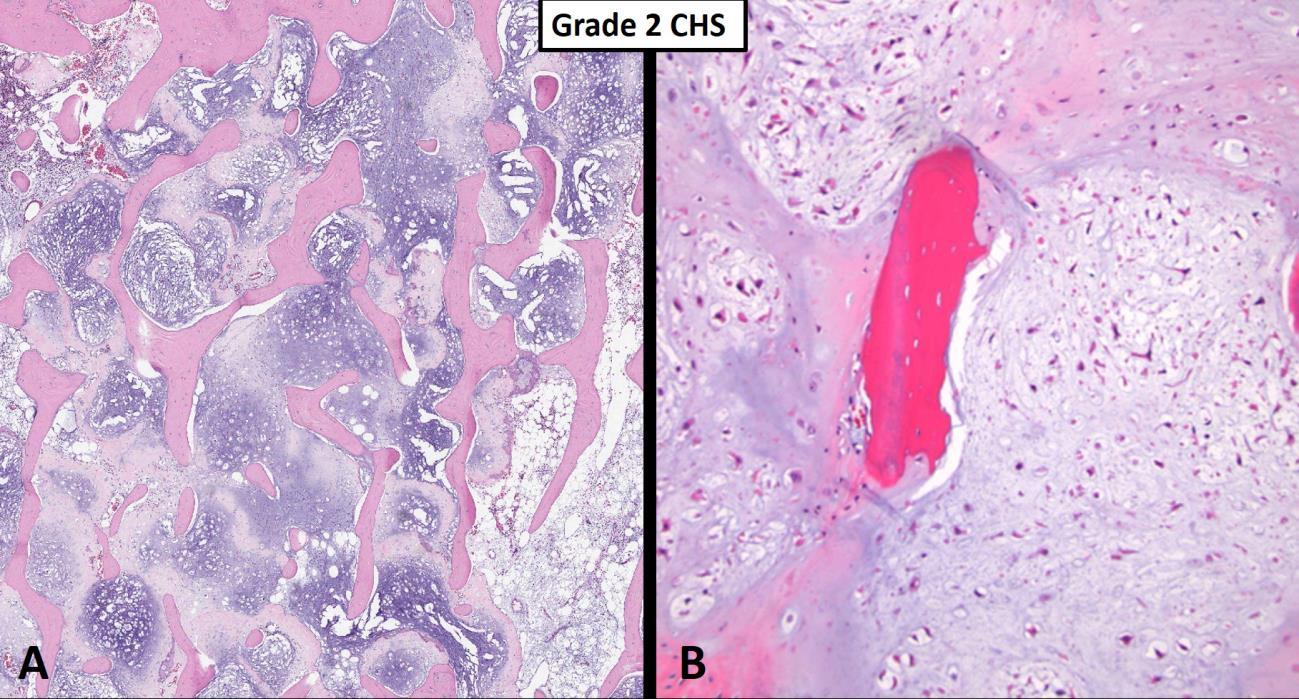
Fig. 3 Grade 2 chondrosarcoma. A Low power view shows a myxoid hyaline cartilage neoplasm completely filling the medullary space, entrapping host bone without obvious remodeling. B Intermediate power images of a grade 2 chondrosarcoma is more cellular than a grade 1 tumor and shows some mild cytologic atypia. Note the imprisonment of native host bone (pink).
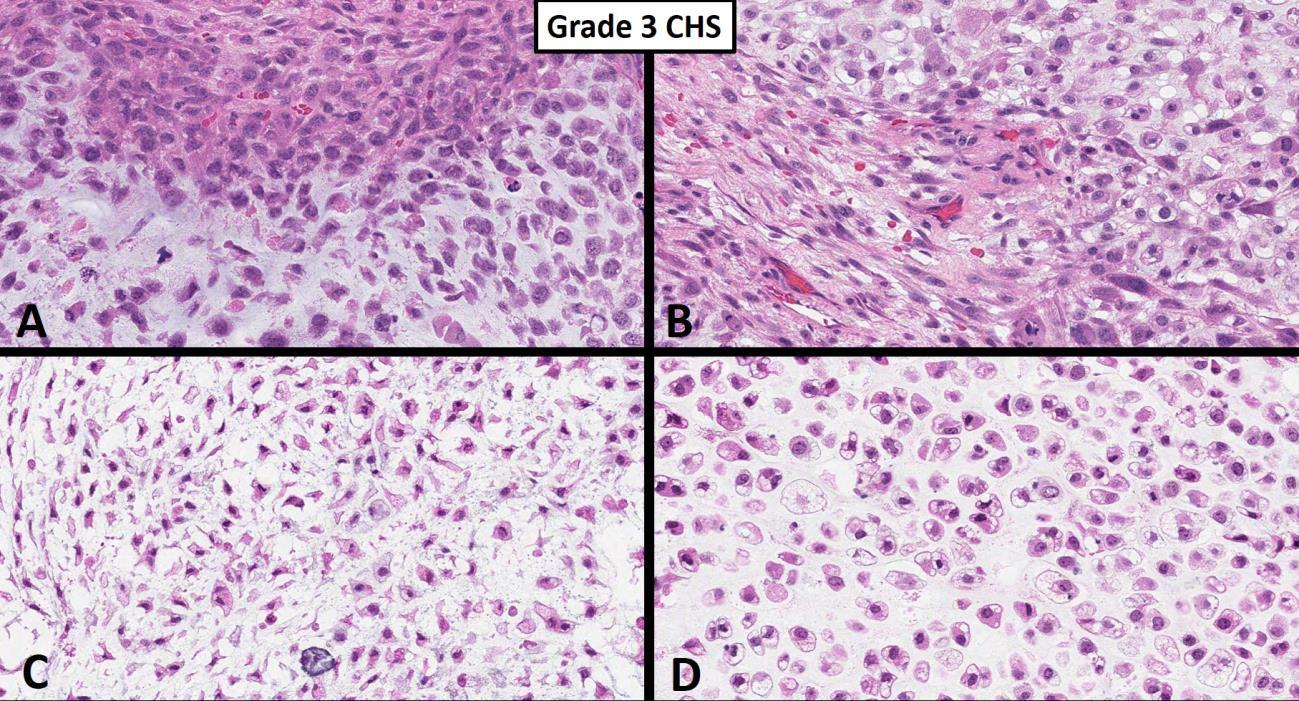
Fig. 4 Grade 3 chondrosarcoma. A-D. High power images show an overtly malignant high-grade chondrosarcoma consisting of pleomorphic chondrocytes with extreme hypercellularity, spindling and rare mitotic activity.
Caveats in Histologic Grading
The grade of chondrosarcoma is an important predictor of behavior and subsequent clinical outcome; however, important caveats need mentioning. First, definitive grading is not always possible on biopsy specimens. Second, histologic grading of chondrosarcoma is subjective as diagnostic criteria are not well established. Third, many chondrosarcomas may progress (transition to a higher but not a lower grade) and, therefore, adequate sampling of a resection specimen by the pathologist is necessary to evaluate for higher grade areas.
Secondary Peripheral Chondrosarcoma
Secondary peripheral chondrosarcoma is an uncommon subtype of conventional chondrosarcoma originating at the surface of the bone in a pre-existing osteochondroma, often in the setting of multiple hereditary exostosis (MHE). Histologic grading and prognosis of secondary peripheral chondrosarcoma is similar to that of primary central chondrosarcoma.3
Periosteal chondrosarcoma
Periosteal chondrosarcoma is a rare chondrosarcoma subtype representing 2.5% of all chondrosarcomas. Periosteal chondrosarcoma occurs in a younger age group with a peak incidence in the third decade of life and, like conventional central chondrosarcoma, has a male predominance.4 Periosteal chondrosarcoma predominantly affects metaphysis (fluted portion) of long bone and the tumor arises on the surface of the cortex generally not involving the medullary canal. Periosteal chondrosarcomas are typically larger than 5.0 cm and there is often invasion of the underlying cortex. In contrast to conventional chondrosarcoma, histologic grading does not predict outcome. Periosteal chondrosarcoma has a relatively low metastatic rate (5-12%) and metastases will generally spread to the lung.5
Chondrosarcoma Variants
The remaining 10-20% of chondrosarcomas comprise a group of variants including clear cell chondrosarcoma, mesenchymal chondrosarcoma and dedifferentiated chondrosarcoma. Of note (and despite its name), extra skeletal myxoid chondrosarcoma is not a true “chondrosarcoma” and is better classified as a translocation sarcoma of unknown histogenesis harboring with the canonical t(9;22) EWSR1-NR4A3 fusion (Fig. 5).6
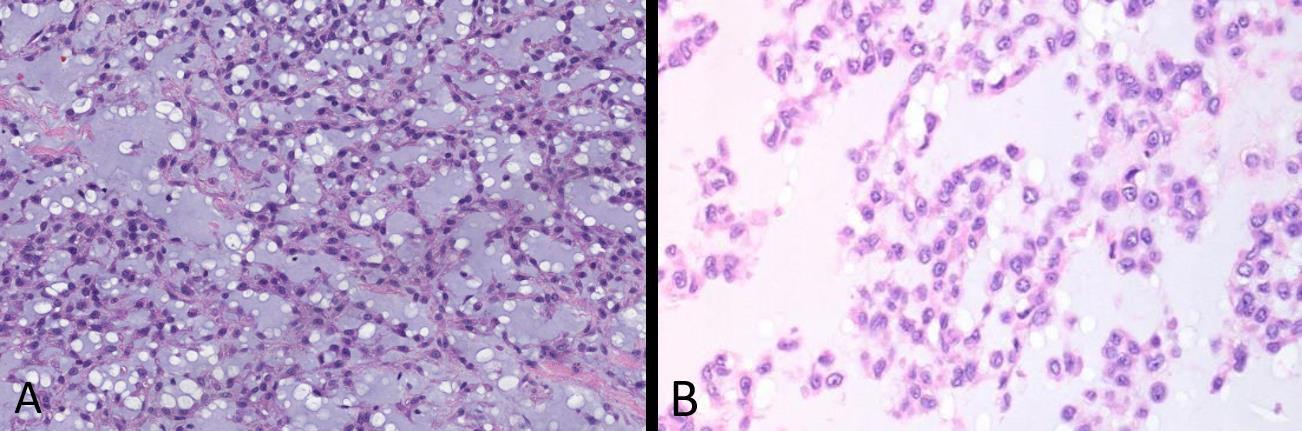
Fig. 5 Extraskeletal myxoid chondrosarcoma A Intermediate power and B high power images of extraskeletal myxoid chondrosarcoma show uniform round cells with scant eosinophilic cytoplasm growing in cords and chains with a chondromyxoid background. This tumor is not a “true” chondrosarcoma, despite its name, and is now known to be a translocation sarcoma, most commonly t(9;22) forming an EWSR1-NR4A3 fusion.
Clear cell chondrosarcoma
Clear cell chondrosarcoma is a low-grade malignant chondrosarcoma generally affecting the epiphysis (ends) of long bones in older adults. The histologic diagnosis of clear cell chondrosarcoma requires the identification of round to polygonal chondrocytes with lightly eosinophilic (pink) to somewhat clear cytoplasm (Fig. 6). A common finding in clear cell chondrosarcoma is the presence of conventional chondrosarcoma, osteoclast-like giant cells and woven bone fragments which, especially on small biopsies, can easily be misinterpreted as chondroblastic osteosarcoma. Clear cell chondrosarcoma rarely metastasizes and has a good overall prognosis. Surgery alone is the mainstay of treatment and the majority of patients can expect to be cured following resection.7,8
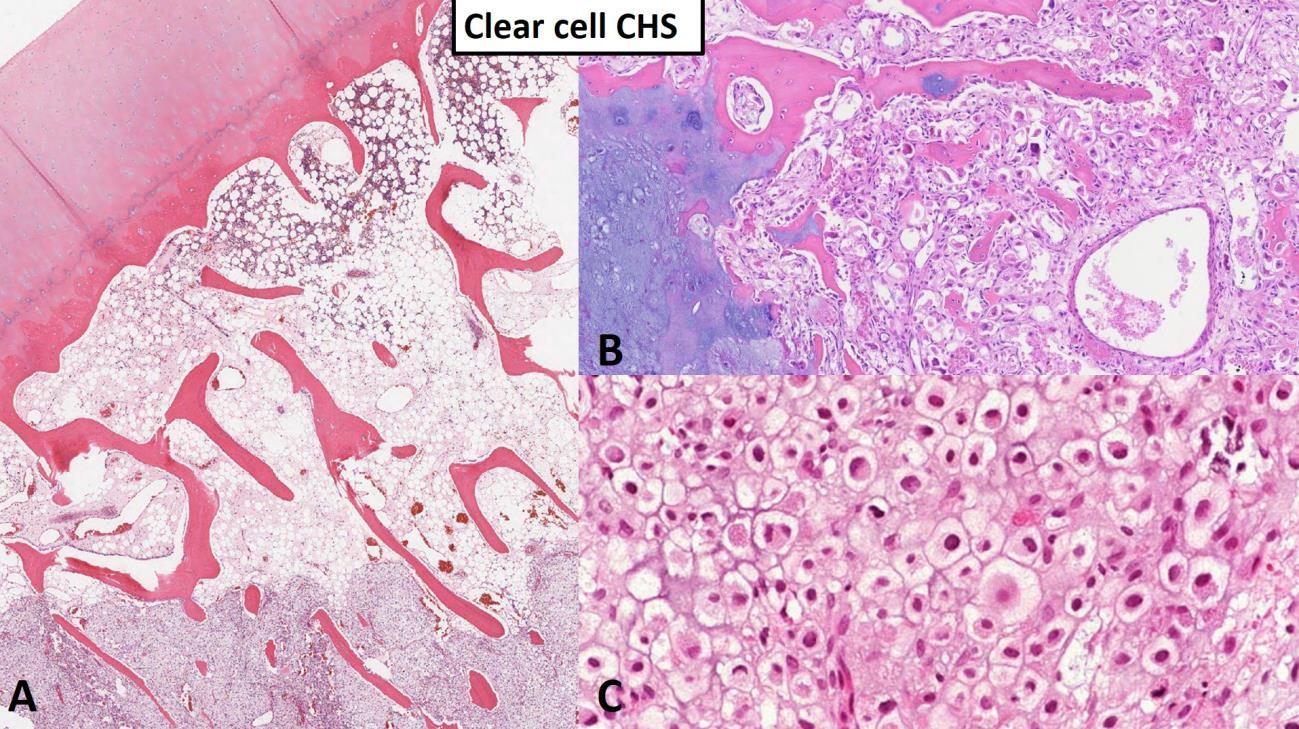
Fig. 6 Clear cell chondrosarcoma. A Low power image of clear cell chondrosarcoma (lower portion of the image) entrapping host bone. Note the articular surface at the top left as clear cell chondrosarcoma originates from the epiphysis of long bone in most cases. B Approximately 50% of clear cell chondrosarcoma contains portions of conventional chondrosarcoma (left portion of image) and small fragments of woven bone (linear pink structures). C High power image shows polygonal tumor cells composed of lightly eosinophilic (pink) to somewhat clear cytoplasm with prominent cytoplasmic membranes and condensed (dark) nuclei.
Mesenchymal chondrosarcoma
Mesenchymal chondrosarcoma is a malignant biphasic neoplasm characterized by a primitive small round cell sarcoma admixed within a low-grade hyaline cartilage tumor (Fig. 7). Mesenchymal chondrosarcoma may affect patients of all age groups but is commonly seen in both children/young adults and older patients. The ribs, skull base and trunk are common locations for mesenchymal chondrosarcoma (Fig. 8); and, unlike other chondrosarcoma subtypes, mesenchymal chondrosarcoma may occur as a primary soft tissue tumor without involvement of bone. Mesenchymal chondrosarcoma contains a disease defining gene translocation, HEY1-NCOA2, which is seen as a fusion on chromosome 8 and is present in > 95% of cases. Mesenchymal chondrosarcoma is an indolent sarcoma with a prolonged survival but a poor long-term outcome due to high probability of late metastasis and ultimate death from disease. One patient reported in the literature died with metastatic mesenchymal chondrosarcoma 23 years after his first operation.9
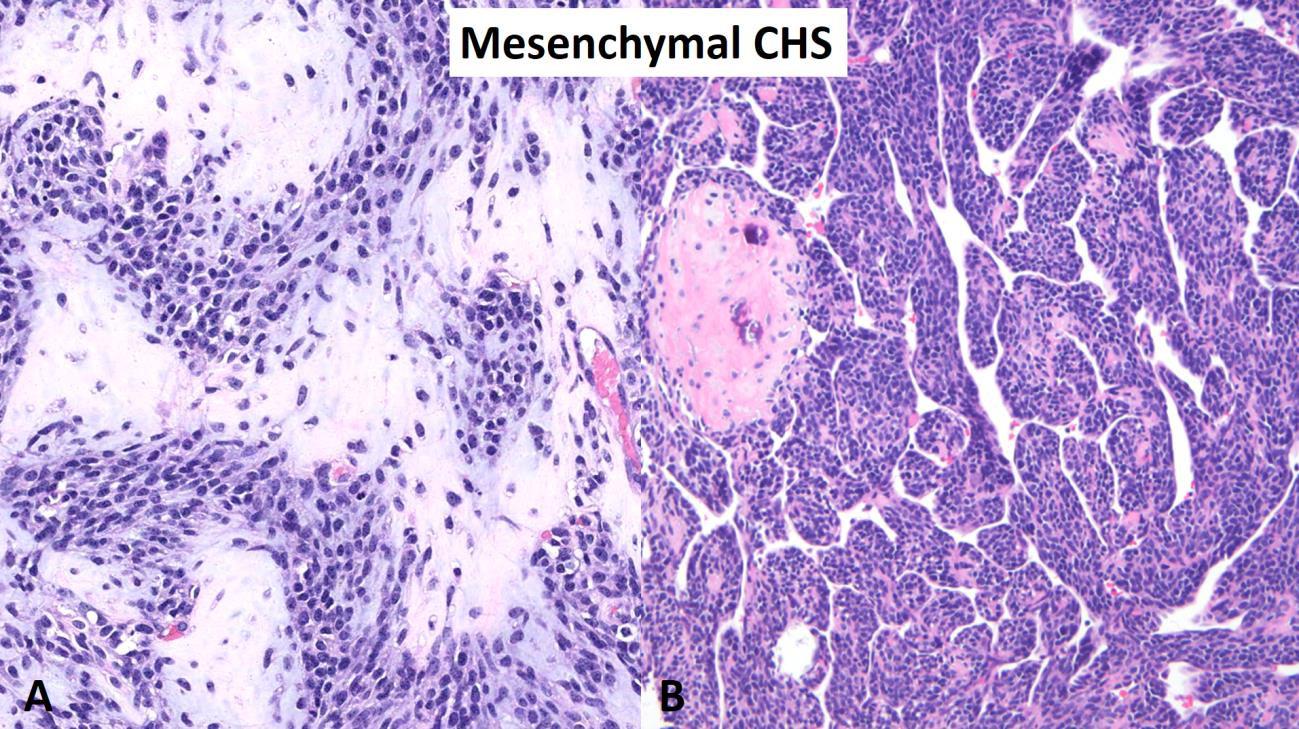
Fig. 7 Mesenchymal chondrosarcoma. A High-power image of mesenchymal chondrosarcoma showing a biphasic neoplasm consisting on a mixture of a small round blue cell tumor and a second component of well-differentiated hyaline cartilage. B Intermediate power photomicrograph showing a predominant small round blue cell tumor with nodules of hyaline cartilage. Note the branching vascular pattern also known as “hemangiopericytoma-like”.
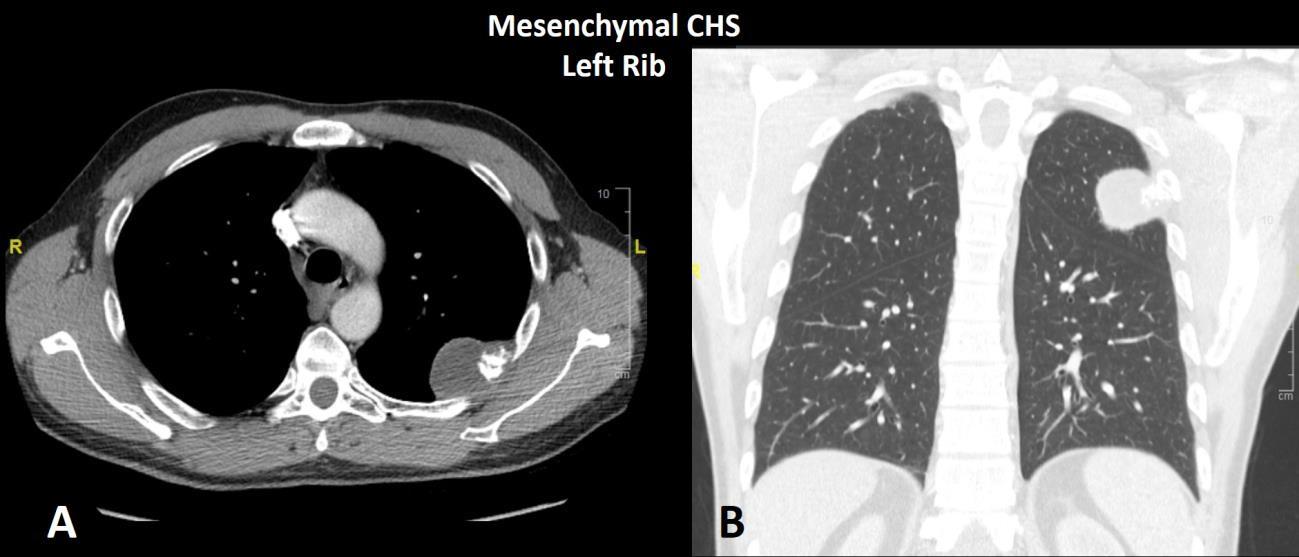
Fig. 8 CT of mesenchymal chondrosarcoma left rib. A Axial and B coronal CT shows a infiltrative neoplasm of the posterior left rib with focal osteochondral matrix production.
Dedifferentiated chondrosarcoma
Dedifferentiated chondrosarcoma is a rare biphasic sarcoma characterized by a low to intermediate grade hyaline cartilage neoplasm component which is juxtaposed with a high-grade malignant sarcoma typically consisting of pleomorphic spindle cells with ample mitotic activity and necrosis (Fig. 9). A helpful diagnostic feature of dedifferentiated chondrosarcoma versus other malignant tumors in the differential diagnosis (high grade chondroblastic osteosarcoma, high-grade chondrosarcoma) is that dedifferentiated chondrosarcoma maintains a separation of the two components without mixing. Radiology is often helpful with the identification of dedifferentiated chondrosarcoma as these tumors may show distinct lower-grade and higher-grade components on advanced imaging (Fig. 10). Dedifferentiated chondrosarcoma has numerous molecular abnormalities including IDH1/2 mutations and a poor overall prognosis with high rates of metastasis and death from disease.10
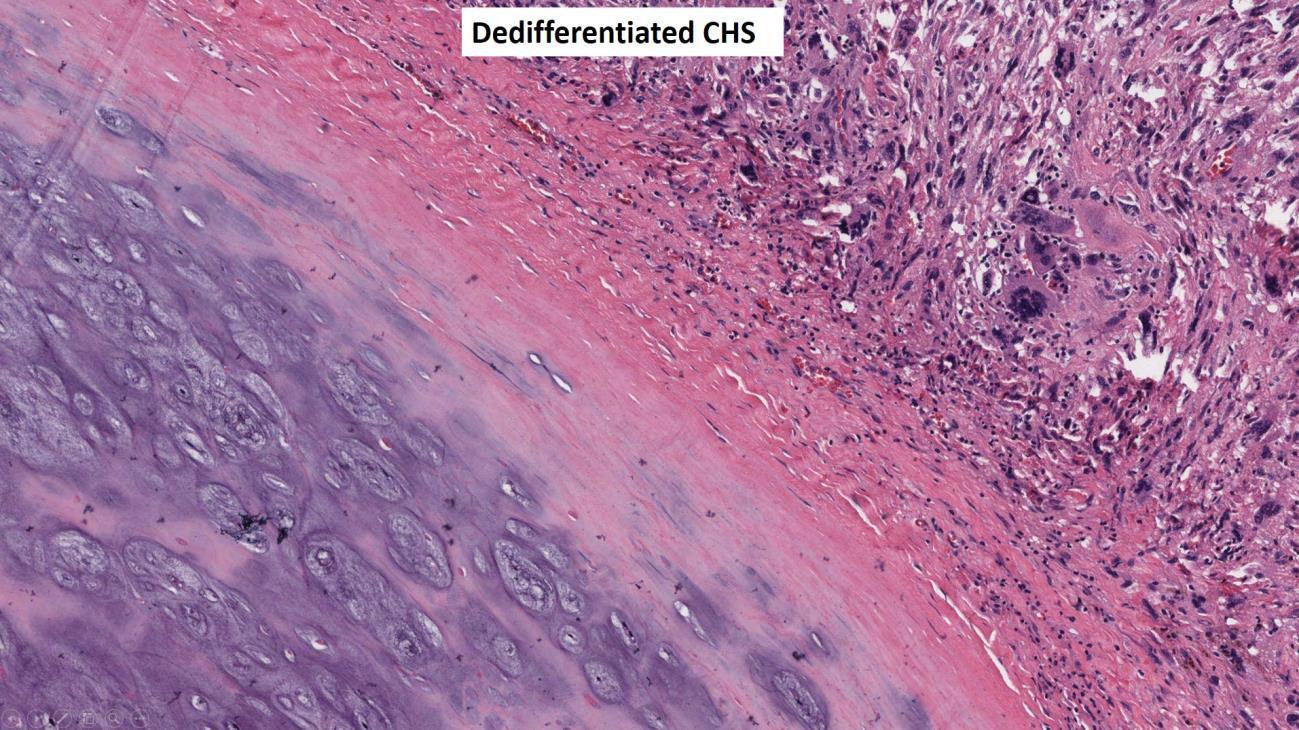
Fig. 9 Dedifferentiated chondrosarcoma. Intermediate power image showing a juxtaposition of two separate components without mixing; hyaline cartilage tumor on the lower left and a high-grade pleomorphic spindle cell sarcoma on the upper right.
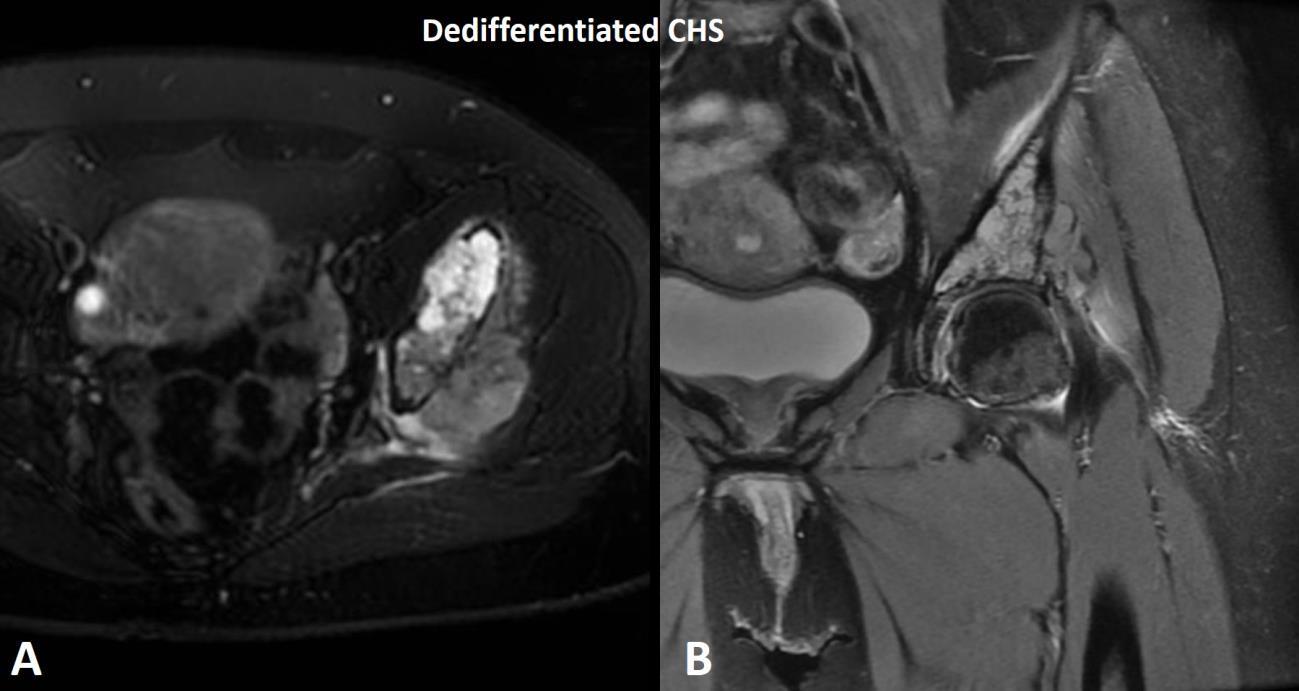
Fig. 10 MRI of dedifferentiated chondrosarcoma. A T2 Axial and B coronal MRI shows a T2 hyperintense (bright) tumor mass representing the low-grade hyaline cartilage component and a T2 hypointense (dark) component representing the high-grade pleomorphic spindle cell sarcoma component of dedifferentiated chondrosarcoma.
Molecular Genetics
While not necessary for the diagnosis in most cases, genetics have been recently applied to further define chondrosarcomas and other tumors in the differential diagnosis, such as chondroblastic osteosarcoma, which has a vastly different treatment approach (osteosarcomas are treated with preoperative chemotherapy). In general, chondrosarcomas can be grouped into four main categories by genetic analysis; those with IDH1/IDH2 gene mutations and secondary “peripheral” chondrosarcomas characterized by mutations in EXT1 /EXT2 genes, chondrosarcomas with recurrent gene translocations such as HEY1-NCOA2 in mesenchymal chondrosarcoma and those without consistent genetic alterations.11
Summary /Conclusions
In summary, chondrosarcoma is the 2nd most common primary bone tumor after osteosarcoma, more commonly affects older patients and has a male predominance. Primary central chondrosarcoma (intramedullary) is by far the most common subtype of conventional chondrosarcoma; however, secondary peripheral chondrosarcoma (tumors arising on the bone surface in the setting of a pre-existing osteochondroma) accounts for approximately 10% of cases. Conventional chondrosarcomas are graded I-III and more than 90% of cases are low to intermediate grade (I-II). Rare variants include the low-grade clear cell chondrosarcoma with a good overall prognosis and aggressive variants including mesenchymal chondrosarcoma (biphasic neoplasm with late metastasis and a poor overall survival) and dedifferentiated chondrosarcoma (biphasic neoplasm with early metastasis and poor overall survival). Radiology is an essential component to the proper diagnosis of cartilage neoplasms as most bone pathologists will not render a final diagnosis until they have reviewed the imaging studies with an experienced musculoskeletal radiologist.
Finally, the treatment of most conventional chondrosarcomas is generally surgery without chemotherapy or radiation. However, innovative approaches towards treating Chondrosarcoma that inhibit and suppress cell growth are being studied. The referral for treatment of Chondrosarcoma should be in a specialized Sarcoma Treatment Center with a multidisciplinary team approach. The Sarcoma Alliance contains a list of these specialized centers. https://sarcomaalliance.org/resources/sarcoma-center/list/ The histologic grade, subtype of chondrosarcoma and the clinical stage (tumor size, location, presence or absence of metastasis) are all necessary constituents to the overall multidisciplinary treatment plan.
REFERENCES
1. WHO Classification of Tumours Editorial Board: Soft tissue and bone tumours: Lyon (France): International Agency for Research on Cancer; 2020. 5th edition; vol 3.
2. Björnsson, J., McLeod, R. A., Unni, K. K., Ilstrup, D. M., & Pritchard, D. J. (1998). Primary chondrosarcoma of long bones and limb girdles. Cancer, 83(10), 2105–2119.
3. Ahmed, A. R., Tan, T.-S., Unni, K. K., Collins, M. S., Wenger, D. E., & Sim, F. H. (2003). Secondary chondrosarcoma in osteochondroma: report of 107 patients. Clinical Orthopaedics and Related Research, 411, 193–206.
4. Picci, P., Manfrini, M., Fabbri, N., Gambarotti, M., & Vanel, D. (2014). Atlas of Musculoskeletal Tumors and Tumorlike Lesions: The Rizzoli Case Archive. Springer Science & Business Media.
5. Cleven, A. H. G., Zwartkruis, E., Hogendoorn, P. C. W., Kroon, H. M., Briaire-de Bruijn, I., & Bovée, J. V. M. G. (2015). Periosteal chondrosarcoma: a histopathological and molecular analysis of a rare chondrosarcoma subtype. Histopathology, 67(4), 483–490.
6. Rubin, B. P., & Fletcher, J. A. (1999). Skeletal and extraskeletal myxoid chondrosarcoma: related or distinct tumors? Advances in Anatomic Pathology, 6(4), 204–212.
7. Unni, K. K., Dahlin, D. C., Beabout, J. W., & Sim, F. H. (1976). Chondrosarcoma: clear-cell variant. A report of sixteen cases. The Journal of Bone and Joint Surgery. American Volume, 58(5), 676– 683.
8. Collins, M. S., Koyama, T., Swee, R. G., & Inwards, C. Y. (2003). Clear cell chondrosarcoma: radiographic, computed tomographic, and magnetic resonance findings in 34 patients with pathologic correlation. Skeletal Radiology, 32(12), 687–694.
9. Salvador, A. H., Beabout, J. W., & Dahlin, D. C. (1971). Mesenchymal chondrosarcoma— observations on 30 new cases. Cancer, 28(3), 605–615.
10. Bovée, J. V., Cleton-Jansen, A. M., Rosenberg, C., Taminiau, A. H., Cornelisse, C. J., & Hogendoorn, P. C. (1999). Molecular genetic characterization of both components of a dedifferentiated chondrosarcoma, with implications for its histogenesis. The Journal of Pathology, 189(4), 454–462.
11. Scotlandi, K., Hattinger, C. M., Pellegrini, E., Gambarotti, M., & Serra, M. (2020). Genomics and Therapeutic Vulnerabilities of Primary Bone Tumors. Cells, 9(4). https://doi.org/10.3390/cells9040968

John M. Gross, MD
John M. Gross, M.D. is a Faculty Assistant professor, bone and soft tissue, surgical pathology at Johns Hopkins University. John M. Gross, MD joined the Chondrosarcoma Foundation Board in May, 2020. During his transition from the Mayo Clinic in Rochester, MN to Johns Hopkins University in Baltimore, MD; Dr. Gross worked on this article in his spare time. It is written to educate Oncologists, Sarcoma Specialists, Oncology Surgeons, Oncology Radiologists, and General Practitioners as well as patients diagnosed with Chondrosarcoma.
Dr. Gross completed his medical degree at Creighton University School of Medicine (Omaha) where he also completed his anatomic and clinical pathology residency followed by fellowships in bone and soft tissue pathology and surgical pathology at the University of Washington (Seattle) and the Mayo Clinic (Rochester, MN). Prior to becoming a pathologist, Dr. Gross completed his internship in general surgery at Cleveland Clinic Florida where he met his wife (Stephanie). Dr. Gross is board certified in anatomic and clinical pathology and an assistant professor of bone and soft tissue pathology and surgical pathology at Johns Hopkins University (Baltimore, MD). His academic interests include bone and soft tissue tumors and molecular oncology.

